Semiconductor clusters
People Involved: I. Abdolhosseini Sarsari, S. J. Hashemifar, H. Salamati
Density functional, full-potential computations are performed to study the origin and consequences of the ring to cage structural crossover in (ZnO)n (n = 2–16) clusters. The origin of this structural crossover, which is found to occur at n = 10, is studied by investigating the behavior of the Zn–O–Zn bond angle. It was argued that the Zn–O–Zn bond angle in the ring (ZnO)9 cluster exceeds its equilibrium value and hence induces a 3D zigzag structural distortion in the system. The stability of the ring structures in the smaller ZnO clusters is attributed to their stronger individual bonds, while the higher number of bonds in the larger clusters enhances the stability of their cage structures.
It is declared that 12 is the lowest magic number of ZnO clusters in the ground state, while finite temperature vibrational excitations enhance the relative stability of the (ZnO)9 cluster to make it a magic system at temperatures above about 170 K.
The obtained electronic structure of the clusters before and after applying the many-body GW corrections evidence a size-induced redshift originating from the ring to cage structural crossover in the system. The behavior of the electron density bond points of the clusters along with the extrapolated cluster binding energy at very large sizes may indicate the existence of a metastable structure for large ZnO nanostructures, different from the bulk ZnO structure.
We argued that the high relative stability of the ring (ZnO)9 cluster at room temperature is due to the thermal activation of the high-energy vibrational modes of the larger cage clusters.
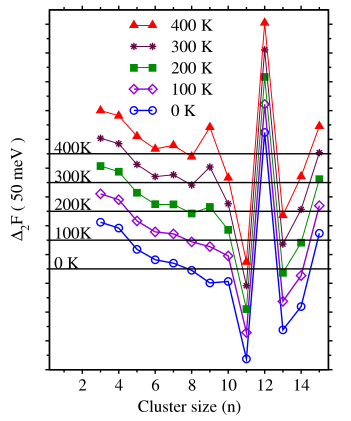
Fig. Second-order difference of the Helmholtz free energy of the ZnO clusters as a function of the cluster size at different temperatures. The horizontal lines show the reference lines at different temperatures.
To learn more:
First-principles study of ring to cage structural crossover in small ZnO Clusters
I. Abdolhosseini Sarsari, S. Javad Hashemifar, Hadi Salamati
J. Phys.: Condens. Matter 24 (2012) 505502 (7pp)